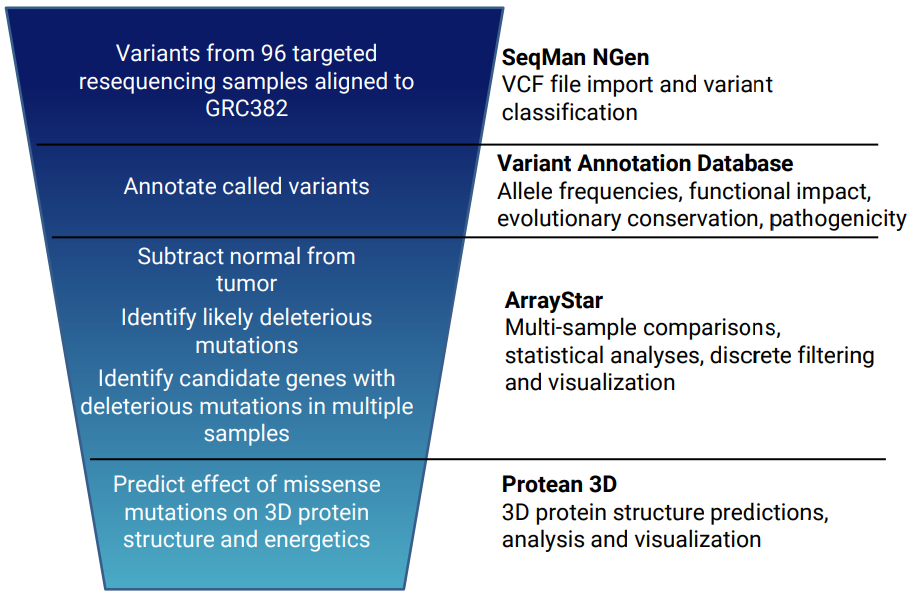
Why is variant annotation important?
Variant analysis can be used to:
Identify the cause(s) of disease by comparing the DNA of affected and unaffected individuals.
Understand evolution by doing phylogenetic comparisons of variants in different populations.
Identify somatic variations that occur in mixed cell populations of mosaic or tumor tissues.
Understand the biology of a mutation by learning how variants affect protein structure and function.
Identify variants in viral or bacterial strains that may affect the duration and intensity of an epidemic.
What type of sequencing data do I need for analyzing variants?
Some common sequencing technologies that create data suitable for variant annotation and analysis include Sanger/ABI, Illumina, Ion Torrent, PacBio and Oxford Nanopore. Sanger technology is still widely used for small scale variant analysis where accuracy is most important, while the Illumina platform provides both accuracy and high throughput variant analysis. Long read platforms have much improved accuracy for variant analysis and the extended read lengths also make additional analysis such has haplotype phasing and large structural variant analysis more practical.
During NGS sequencing for Illumina and Ion Torrent, the pipeline tools associated with the sequencing instrument usually clean up the data files. This is normally sufficient, but some output sequence files can benefit from scanning with a third-party tool like FastQC.
By contrast, Sanger data usually contains many base calling errors at the 5’ and 3’ ends where the chromatogram peaks are not high quality. This type of data requires a high-quality software program that can accurately trim the sequence ends prior to assembly.
What should I look for in a variant analysis software package?
To analyze variant data, you will first need software to align the experimental sequences against a known reference sequence. Reference sequences for common model organisms are available for free download at sites like NCBI.
Why is the filtering step important?
Identifying more interesting variations from thousands of located variants can be challenging and often requires several rounds of analysis and data filtering.
Most variants are benign mutations in the DNA that do not affect protein coding and are not located in or near genes. Default filters should be in place to remove these variations from the initial analysis. Variant filtering can quickly eliminate thousands of variants from consideration and save you time and frustration.
What are some of the biggest challenges in variant annotation?
Most researchers begin by using existing annotation from the reference genome to identify variants in coding regions whose presence affects protein coding (AKA “non-synonymous” variants). Some organisms may also have sets of previously identified variants, usually in VCF file format. This annotation information can be imported into the analysis so the researcher can differentiate between previously characterized variants and novel variations.
Annotations for known variants may include information on allele frequency distribution, as well as references or links to additional annotation databases that can be used to interpret the functional impact of the variation.
Online variant annotation databases contain massive amounts of useful information but accessing this data can be challenging. Most databases are prohibitively large for manual searches. In addition, many use proprietary formats that require researchers to pay for access. For these reasons, incorporating variant annotations from multiple sources can be extremely unwieldy and time-consuming for researchers working with even small data sets.
Even with initial filtering, there can be thousands of variants remaining. The ability to apply additional filtering on the imported variant database annotations is critical to creating a manageable and meaningful data set for interpretation.