
The Advantages and Challenges of AlphaFold 2 and AlphaFold-Multimer
This chapter will discuss the advantages and challenges of using the open-source version of AlphaFold 2 and will describe how NovaFold AI can help streamline this workflow.
What is special about AlphaFold 2 and AlphaFold Multimer?
AlphaFold 2 was developed by DeepMind, a subsidiary of Alphabet Inc. (Google). This unique algorithm uses artificial intelligence/machine-learning to calculate the likely 3D structure of a protein.

Over the past eight CASP experiments, just three groups have attained first place in the competition, based on combined group z-scores for “regular” targets. (Table 1).
| Event | Year | Winner |
| CASP7 | 2006 | Zhang |
| CASP8 | 2008 | Zhang |
| CASP9 | 2010 | Zhang |
| CASP10 | 2012 | Zhang |
| CASP11 | 2014 | Zhang |
| CASP12 | 2016 | Baker (Rosetta group) |
| CASP13 | 2018 | A7D |
| CASP14 | 2020 | AlphaFold 2 |
| CASP15 | 2022 | Yang-Server (AlphaFold 2 did not compete) |
Table 1. Highest-ranked algorithm in each CASP experiment from 2006-present based on combined group z-scores for regular targets. Color is used to denote algorithms from the same group. AlphaFold 2 is an updated iteration of the “A7D” algorithm. “Zhang” is another name for the I-TASSER algorithm used by DNASTAR NovaFold, while “Yang-Server” is a different algorithm from the same group.
I-TASSER (called Zhang or Yang-Server in the CASP events) has won more CASP experiments than any other algorithm and forms the basis of DNASTAR’s NovaFold application.
A turning point came with the results of CASP14 in 2020. AlphaFold 2 made a splash in the protein research community when it beat all 145 other algorithms as a first-time CASP competitor. Not only did AlphaFold 2 win this global experiment, but it was 2.65 times more accurate than its closest rival, the Baker (Rosetta Group) algorithm that had won CASP13 (Figure 1).
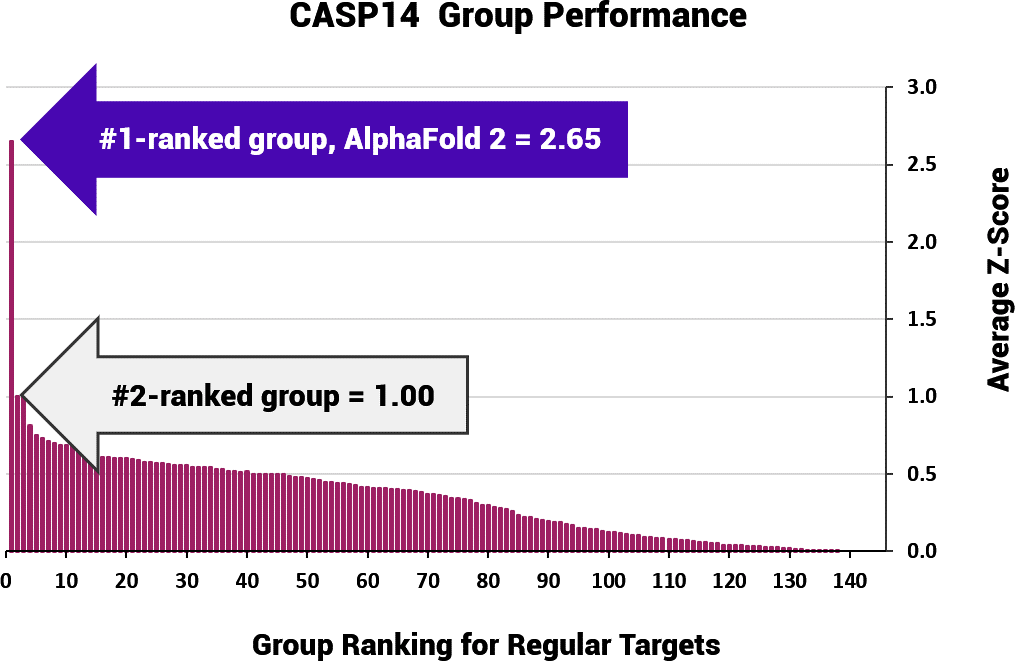
Figure 1. In the CASP14 Challenge, AlphaFold 2 achieved an average z-score* of 2.65 over 92 “regular” targets. The closest competitor achieved only 1.00 for the same number of targets (Graph created from this table).
* The adjusted z-score, essentially the number of standard deviations (SD) above the mean of the full set of models, has been computed in recent CASP experiments using the protocol from Croll TI, Sammito MD et al. Evaluation of template-based modeling in CASP13, Proteins, Volume 87, Issue 12, Pages 1113-1127.
Based on the CASP results and the experiences of real users worldwide, AlphaFold 2 is the most accurate and the fastest algorithm available for single-chain proteins, finishing most structure predictions in a few hours or less. AlphaFold 2 can also predict the 3D structures of very challenging proteins like:
- Membrane-bound proteins
- Fusion proteins
- Cytosolic domains (CDs)
- Extra-cellular regions
- G-protein-couple receptors (GPCRs)
The algorithm can also model multiple domains and their interactions with linkers, also known as multidomain protein structure prediction.
AlphaFold-Multimer is an extension of the AlphaFold 2 algorithm and was unveiled by DeepMind in 2021. Compared to AlphaFold 2, AlphaFold-Multimer uses two additional metrics for evaluating prediction accuracy. To learn more about these, see this EMBL-EBI training page.
Unlike single-chain prediction algorithms, which can be objectively tested in the CASP experiments, there is no similar test (so far) for multi-chain structure prediction. Nevertheless, AlphaFold-Multimer is believed to be the most accurate algorithm to date for predicting the quaternary structures of protein-protein complexes.
To learn more about the AlphaFold2 algorithm, see Jumper, J, Evans, R, Pritzel, A et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). To learn more about AlphaFold-Multimer, see Evans R, O’Neill, M et al. (2021). Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021 (preprint)
Accessing AlphaFold 2 models for free
The AlphaFold 2 database at EMBL-EBI has 200 million structure predictions that can be downloaded for free. It is definitely worth checking to see if a predicted model for your protein of interest is available in the database.
Still, there are a large number of proteins that have not and will not be predicted and placed in the database. That is because the EMBL-EBI database contains only naturally occurring proteins, and only a subset of those. The database does not include custom fusion proteins, synthetic proteins, and mutations of naturally occurring proteins. In addition, the database contains only one predicted structure per protein, and does not contain alternate splicing versions or isoforms.
Because of these limitations, if you wish to learn about your own protein of interest, you will likely need to set up and run a novel prediction. This workflow is described in the next section.

Using open source AlphaFold 2 and AlphaFold-Multimer to predict novel structures
AlphaFold 2 and AlphaFold-Multimer are freely available to download and use but come with many of the inherent disadvantages of open-source software described in the previous chapter. As with other open-source prediction software, they generally require the assistance of an IT team to set up and are run by typing complex commands into a command line (see Table 2).
In addition, these algorithms run on a local machine, rather than on the cloud, and must be installed on an extremely high-powered and high-capacity Linux computer. For example, AlphaFold 2 and its associated template library—vital for the sequence searching and alignment phase of each prediction—require 2.5 Terabytes of disk space. For most protein researchers, it is impractical to purchase an expensive Linux machine solely to use for a single application.
A final consideration is that open-source versions of these algorithms do not include a protein structure viewer. So, you will need to install and learn additional software in order to view the predicted protein models.
If you already have a Linux computer with Docker installed, and are comfortable using command- line scripts, there are two other alternatives available:
For AlphaFold 2, there is an AlphaFold Colab that runs on the cloud and requires much less disk space than the full AlphaFold 2 installation described above. However, this solution uses a simplified version of AlphaFold 2 that has lower accuracy than the full version and does not work for multi- chain protein molecules (multimers).
AlphaFold-Multimer can be run via ColabFold, which operates within the constraints of Google Colab notebook. However, the Google Colab notebook presents memory limitations that can pose challenges when running the AlphaFold-Multimer algorithm.